
Region and Locset Filters#
This notebook is intentionally small-step: prepare a few morphology presets, define a small set of visualization knobs, then use short calls to show what each filter expression selects.
The most important split is:
Region filters select branch intervals and are used by
Cell.paint(...).Locset filters select branch-local points and are used by
Cell.place(...).
Before the filters themselves, we first make the visualization level and CV policy explicit so every plot is easy to adjust.
import os
os.environ.setdefault('JAX_PLATFORMS', 'cpu')
os.environ.setdefault('PYVISTA_OFF_SCREEN', 'true')
os.environ.setdefault('MPLBACKEND', 'Agg')
from collections import Counter
from pathlib import Path
import warnings
import matplotlib.pyplot as plt
import brainunit as u
import braincell
import braincell.mech as mech
from braincell import Morphology, Cell, CVPerBranch
from braincell.filter import (
AllRegion,
EmptyRegion,
BranchSlice,
BranchInFilter,
BranchRangeFilter,
AtLocation,
RootLocation,
BranchPoints,
Terminals,
UniformSamples,
RandomSamples,
at,
branch_in,
branch_range,
)
warnings.filterwarnings(
'ignore',
message=r'from_points\(\) produced .* zero-length segment',
)
plt.rcParams['figure.dpi'] = 120
1. Prepare Morphology Presets#
The examples move from a tiny tree to richer real morphologies. Later code cells only refer to these variables.
repo_root = Path(braincell.__file__).resolve().parents[1]
morpho_dir = repo_root / 'data' / 'morphology'
morph_simple = Morphology.from_swc(morpho_dir / 'example_tree.swc')
morph_io = Morphology.from_swc(morpho_dir / 'io.swc')
morph_pc = Morphology.from_asc(morpho_dir / 'pc.asc')
MORPH_PRESETS = {
'morph_simple': morph_simple,
'morph_io': morph_io,
'morph_pc': morph_pc,
}
def describe_morph(name, morph):
type_count = dict(sorted(Counter(branch.type for branch in morph.branches).items()))
print(f'{name:<12} branches={len(morph.branches):>4} root={morph.root.name:<10} types={type_count}')
for name, morph in MORPH_PRESETS.items():
describe_morph(name, morph)
Warning: no DISPLAY environment variable.
--No graphics will be displayed.
morph_simple branches= 9 root=soma types={'axon': 2, 'basal_dendrite': 6, 'soma': 1}
morph_io branches= 31 root=soma types={'basal_dendrite': 25, 'soma': 6}
morph_pc branches= 461 root=soma types={'dendrite': 460, 'soma': 1}
2. Visualization Knobs and Display Helpers#
Cell.vis_topology(...) can render the same selection at three topology levels:
level='node': runtime point-tree view. Attachment points, CV midpoints, and branch terminal points can all be visible.level='cv': one node per control volume. This is the default for region examples in this notebook because it shows partial branch coverage clearly.level='branch': one node per morphology branch. This is compact and useful for branch metadata filters. Locsets cannot be drawn at branch level.
The CV view depends on the control-volume policy. Here the global default is CVPerBranch(cv_per_branch=2), meaning each branch is split into two CVs for the visualization cell. Change the constants below and rerun later cells to compare different settings.
Each compare_regions(...) / compare_locsets(...) call also accepts per-panel overrides, so one row can compare different level, layout, cv_per_branch, or even different morphologies.
CELL_CACHE = {}
MORPH_NAMES = {id(morph): name for name, morph in MORPH_PRESETS.items()}
COLORS = ('#ef4444', '#0ea5e9', '#22c55e', '#f59e0b', '#a855f7', '#14b8a6')
# Global plot defaults. Change these and rerun later cells when comparing views.
DEFAULT_MORPH = morph_simple
DEFAULT_REGION_LEVEL = 'cv'
DEFAULT_REGION_LAYOUT = 'twopi'
DEFAULT_LOCSET_LEVEL = 'node'
DEFAULT_LOCSET_LAYOUT = 'kamada_kawai'
DEFAULT_CV_PER_BRANCH = 2
DEFAULT_LAYOUT_SCALE = 1.5
DEFAULT_PANEL_WIDTH = 3.8
DEFAULT_PANEL_HEIGHT = 3.4
def morph_name(morph):
return MORPH_NAMES.get(id(morph), 'morph')
def resolve_morph(morph):
if morph is None:
return DEFAULT_MORPH
if isinstance(morph, str):
return MORPH_PRESETS[morph]
return morph
def get_cell(morph, *, cv_per_branch=None):
cv_per_branch = DEFAULT_CV_PER_BRANCH if cv_per_branch is None else cv_per_branch
key = (id(morph), int(cv_per_branch))
if key not in CELL_CACHE:
cell = Cell(morph, cv_policy=CVPerBranch(cv_per_branch=int(cv_per_branch)))
cell.init_state()
CELL_CACHE[key] = cell
return CELL_CACHE[key]
def _spec_from_item(item, *, default_morph, default_level, default_layout, default_cv_per_branch, default_layout_scale):
if isinstance(item, dict):
spec = dict(item)
else:
label, expr = item
spec = {'label': label, 'expr': expr}
spec['morph'] = resolve_morph(spec.get('morph', default_morph))
spec['level'] = spec.get('level', default_level)
spec['layout'] = spec.get('layout', default_layout)
spec['preset'] = spec.get('preset', None)
spec['layout_scale'] = spec.get('layout_scale', default_layout_scale)
spec['cv_per_branch'] = spec.get('cv_per_branch', default_cv_per_branch)
return spec
def _topology_kwargs(spec, *, ax, highlight_color):
kwargs = {
'level': spec['level'],
'highlight_color': highlight_color,
'node_color': '#d1d5db',
'edge_color': '#e5e7eb',
'root_color': '#000000',
'ax': ax,
'show': False,
}
if spec.get('layout') is not None:
kwargs['layout'] = spec['layout']
if spec.get('preset') is not None:
kwargs['preset'] = spec['preset']
if spec.get('layout_scale') is not None:
kwargs['layout_scale'] = spec['layout_scale']
return kwargs
def _plot_title(spec):
return f"{spec['label']}\n{morph_name(spec['morph'])} | {spec['level']} | cv={spec['cv_per_branch']}"
def compare_regions(
morph=None,
labeled_exprs=(),
*,
level=None,
layout=None,
cv_per_branch=None,
layout_scale=None,
width=None,
height=None,
):
base_morph = resolve_morph(morph)
base_level = DEFAULT_REGION_LEVEL if level is None else level
base_layout = DEFAULT_REGION_LAYOUT if layout is None else layout
base_cv_per_branch = DEFAULT_CV_PER_BRANCH if cv_per_branch is None else cv_per_branch
base_layout_scale = DEFAULT_LAYOUT_SCALE if layout_scale is None else layout_scale
specs = [
_spec_from_item(
item,
default_morph=base_morph,
default_level=base_level,
default_layout=base_layout,
default_cv_per_branch=base_cv_per_branch,
default_layout_scale=base_layout_scale,
)
for item in labeled_exprs
]
width = DEFAULT_PANEL_WIDTH if width is None else width
height = DEFAULT_PANEL_HEIGHT if height is None else height
fig, axes = plt.subplots(1, len(specs), figsize=(width * len(specs), height))
axes = [axes] if len(specs) == 1 else list(axes)
print(
'compare_regions defaults:',
f'morph={morph_name(base_morph)}',
f'level={base_level}',
f'layout={base_layout}',
f'cv_per_branch={base_cv_per_branch}',
)
for index, spec in enumerate(specs):
spec_morph = spec['morph']
expr = spec['expr']
mask = spec_morph.select(expr)
print(
f" {spec['label']:<28}",
f"morph={morph_name(spec_morph):<12}",
f"level={spec['level']:<6}",
f"layout={str(spec['layout'] or spec['preset']):<12}",
f"cv_per_branch={spec['cv_per_branch']:<2}",
f"intervals={len(mask.intervals):>4}",
f"sample={mask.intervals[:4]}",
)
cell = get_cell(spec_morph, cv_per_branch=spec['cv_per_branch'])
kwargs = _topology_kwargs(spec, ax=axes[index], highlight_color=COLORS[index % len(COLORS)])
kwargs['region'] = expr
kwargs['coverage_mode'] = spec.get('coverage_mode', 'fraction')
cell.vis_topology(**kwargs)
axes[index].set_title(_plot_title(spec), fontsize=9)
plt.tight_layout()
plt.show()
def compare_locsets(
morph=None,
labeled_exprs=(),
*,
level=None,
layout=None,
cv_per_branch=None,
layout_scale=None,
width=None,
height=None,
):
base_morph = resolve_morph(morph)
base_level = DEFAULT_LOCSET_LEVEL if level is None else level
base_layout = DEFAULT_LOCSET_LAYOUT if layout is None else layout
base_cv_per_branch = DEFAULT_CV_PER_BRANCH if cv_per_branch is None else cv_per_branch
base_layout_scale = DEFAULT_LAYOUT_SCALE if layout_scale is None else layout_scale
specs = [
_spec_from_item(
item,
default_morph=base_morph,
default_level=base_level,
default_layout=base_layout,
default_cv_per_branch=base_cv_per_branch,
default_layout_scale=base_layout_scale,
)
for item in labeled_exprs
]
width = DEFAULT_PANEL_WIDTH if width is None else width
height = DEFAULT_PANEL_HEIGHT if height is None else height
fig, axes = plt.subplots(1, len(specs), figsize=(width * len(specs), height))
axes = [axes] if len(specs) == 1 else list(axes)
print(
'compare_locsets defaults:',
f'morph={morph_name(base_morph)}',
f'level={base_level}',
f'layout={base_layout}',
f'cv_per_branch={base_cv_per_branch}',
)
for index, spec in enumerate(specs):
if spec['level'] == 'branch':
raise ValueError("Locsets can be visualized with level='node' or level='cv', not level='branch'.")
spec_morph = spec['morph']
expr = spec['expr']
mask = spec_morph.select(expr)
print(
f" {spec['label']:<28}",
f"morph={morph_name(spec_morph):<12}",
f"level={spec['level']:<6}",
f"layout={str(spec['layout'] or spec['preset']):<12}",
f"cv_per_branch={spec['cv_per_branch']:<2}",
f"points={len(mask.points):>4}",
f"sample={mask.display_names[:4]}",
)
cell = get_cell(spec_morph, cv_per_branch=spec['cv_per_branch'])
kwargs = _topology_kwargs(spec, ax=axes[index], highlight_color=COLORS[index % len(COLORS)])
kwargs['locset'] = expr
cell.vis_topology(**kwargs)
axes[index].set_title(_plot_title(spec), fontsize=9)
plt.tight_layout()
plt.show()
2.1 One Region, Different Views#
This first example keeps the filter fixed and changes the display settings. It shows why level and cv_per_branch matter before we start comparing filter logic.
same_region = BranchSlice(branch_index=3, prox=0.10, dist=0.80)
compare_regions(
morph_simple,
[
{'label': 'node view', 'expr': same_region, 'level': 'node', 'preset': 'dendrotweaks', 'layout': None},
{'label': 'cv, 1 per branch', 'expr': same_region, 'level': 'cv', 'layout': 'twopi', 'cv_per_branch': 1},
{'label': 'cv, 2 per branch', 'expr': same_region, 'level': 'cv', 'layout': 'twopi', 'cv_per_branch': 2},
{'label': 'branch view', 'expr': same_region, 'level': 'branch', 'layout': 'dot'},
],
)
compare_regions defaults: morph=morph_simple level=cv layout=twopi cv_per_branch=2
node view morph=morph_simple level=node layout=dendrotweaks cv_per_branch=2 intervals= 1 sample=((3, 0.1, 0.8),)
cv, 1 per branch morph=morph_simple level=cv layout=twopi cv_per_branch=1 intervals= 1 sample=((3, 0.1, 0.8),)
cv, 2 per branch morph=morph_simple level=cv layout=twopi cv_per_branch=2 intervals= 1 sample=((3, 0.1, 0.8),)
branch view morph=morph_simple level=branch layout=dot cv_per_branch=2 intervals= 1 sample=((3, 0.1, 0.8),)
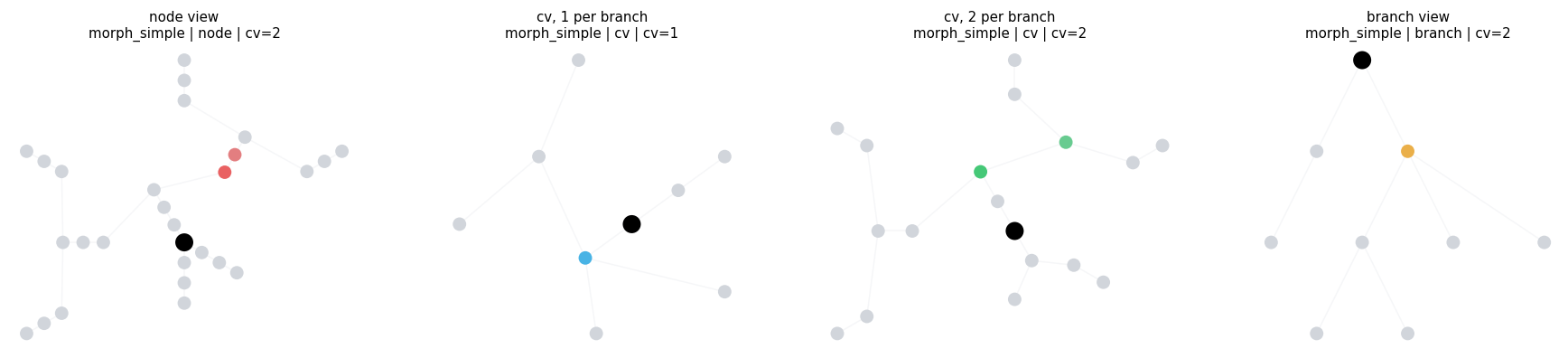
3. Region Filters#
A region is a set of continuous branch intervals: (branch_id, prox, dist).
Start with the small morphology so every selection is easy to inspect.
3.1 Explicit Regions#
Use AllRegion, EmptyRegion, and BranchSlice when you already know the branch ids and normalized coordinates.
compare_regions(
morph_simple,
[
('all branches', AllRegion()),
('branch 3: 10%-80%', BranchSlice(branch_index=3, prox=0.10, dist=0.80)),
('two branch slices', BranchSlice(branch_index=[1, 3], prox=[0.0, 0.20], dist=[1.0, 0.80])),
('empty', EmptyRegion()),
],
level='cv',
layout='twopi',
cv_per_branch=1,
)
compare_regions defaults: morph=morph_simple level=cv layout=twopi cv_per_branch=1
all branches morph=morph_simple level=cv layout=twopi cv_per_branch=1 intervals= 9 sample=((0, 0.0, 1.0), (1, 0.0, 1.0), (2, 0.0, 1.0), (3, 0.0, 1.0))
branch 3: 10%-80% morph=morph_simple level=cv layout=twopi cv_per_branch=1 intervals= 1 sample=((3, 0.1, 0.8),)
two branch slices morph=morph_simple level=cv layout=twopi cv_per_branch=1 intervals= 2 sample=((1, 0.0, 1.0), (3, 0.2, 0.8))
empty morph=morph_simple level=cv layout=twopi cv_per_branch=1 intervals= 0 sample=()
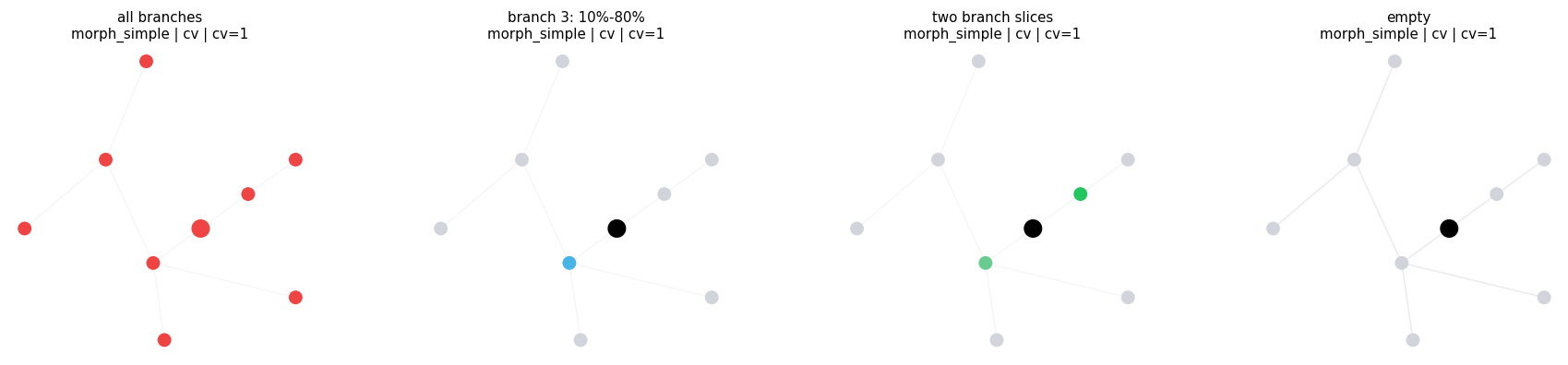
3.2 Branch Metadata Predicates#
Use branch_in(...) when the rule is equality or membership over branch metadata / topology.
compare_regions(
morph_simple,
[
('type == soma', branch_in('type', 'soma')),
('type == basal', branch_in('type', 'basal_dendrite')),
('children of branch 3', branch_in('parent_id', {3})),
('order in {2, 3}', branch_in('branch_order', {2, 3})),
],
level='branch',
layout='dot',
)
compare_regions defaults: morph=morph_simple level=branch layout=dot cv_per_branch=2
type == soma morph=morph_simple level=branch layout=dot cv_per_branch=2 intervals= 1 sample=((0, 0.0, 1.0),)
type == basal morph=morph_simple level=branch layout=dot cv_per_branch=2 intervals= 6 sample=((3, 0.0, 1.0), (4, 0.0, 1.0), (5, 0.0, 1.0), (6, 0.0, 1.0))
children of branch 3 morph=morph_simple level=branch layout=dot cv_per_branch=2 intervals= 3 sample=((4, 0.0, 1.0), (7, 0.0, 1.0), (8, 0.0, 1.0))
order in {2, 3} morph=morph_simple level=branch layout=dot cv_per_branch=2 intervals= 6 sample=((2, 0.0, 1.0), (4, 0.0, 1.0), (5, 0.0, 1.0), (6, 0.0, 1.0))
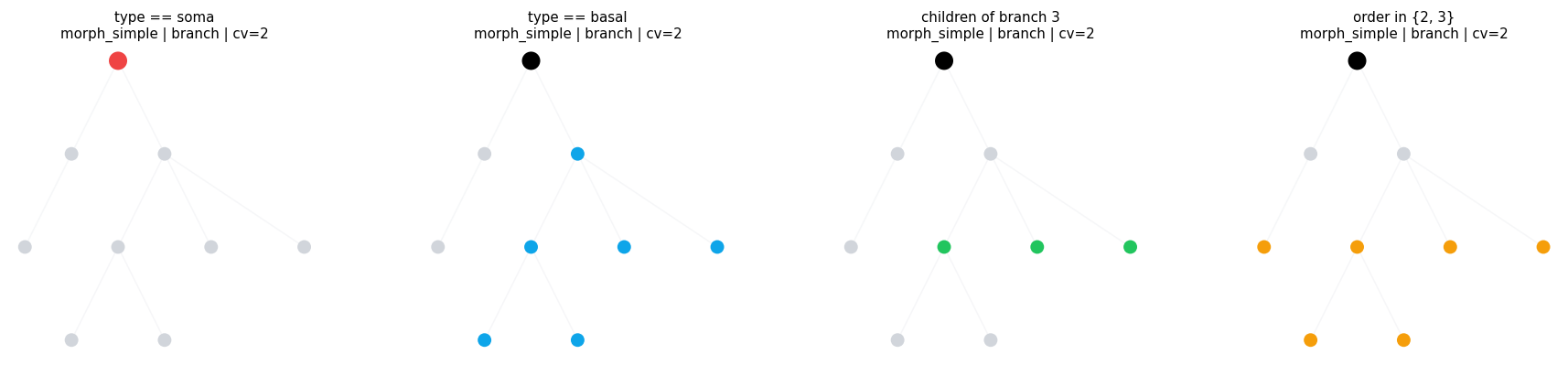
3.3 Numeric and Quantity Ranges#
Use branch_range(...) for numeric topology values and unit-carrying branch metrics.
compare_regions(
morph_simple,
[
('branch_id [1, 4]', branch_range('branch_id', (1, 4), closed='both')),
('length >= 20 um', branch_range('length', (20.0 * u.um, None), closed='left')),
('mean_radius <= 1 um', branch_range('mean_radius', (None, 1.0 * u.um), closed='right')),
('volume <= 80 um^3', branch_range('volume', (None, 80.0 * (u.um ** 3)), closed='right')),
],
level='branch',
layout='dot',
)
compare_regions defaults: morph=morph_simple level=branch layout=dot cv_per_branch=2
branch_id [1, 4] morph=morph_simple level=branch layout=dot cv_per_branch=2 intervals= 4 sample=((1, 0.0, 1.0), (2, 0.0, 1.0), (3, 0.0, 1.0), (4, 0.0, 1.0))
length >= 20 um morph=morph_simple level=branch layout=dot cv_per_branch=2 intervals= 5 sample=((1, 0.0, 1.0), (3, 0.0, 1.0), (4, 0.0, 1.0), (5, 0.0, 1.0))
mean_radius <= 1 um morph=morph_simple level=branch layout=dot cv_per_branch=2 intervals= 3 sample=((1, 0.0, 1.0), (2, 0.0, 1.0), (8, 0.0, 1.0))
volume <= 80 um^3 morph=morph_simple level=branch layout=dot cv_per_branch=2 intervals= 3 sample=((2, 0.0, 1.0), (6, 0.0, 1.0), (8, 0.0, 1.0))
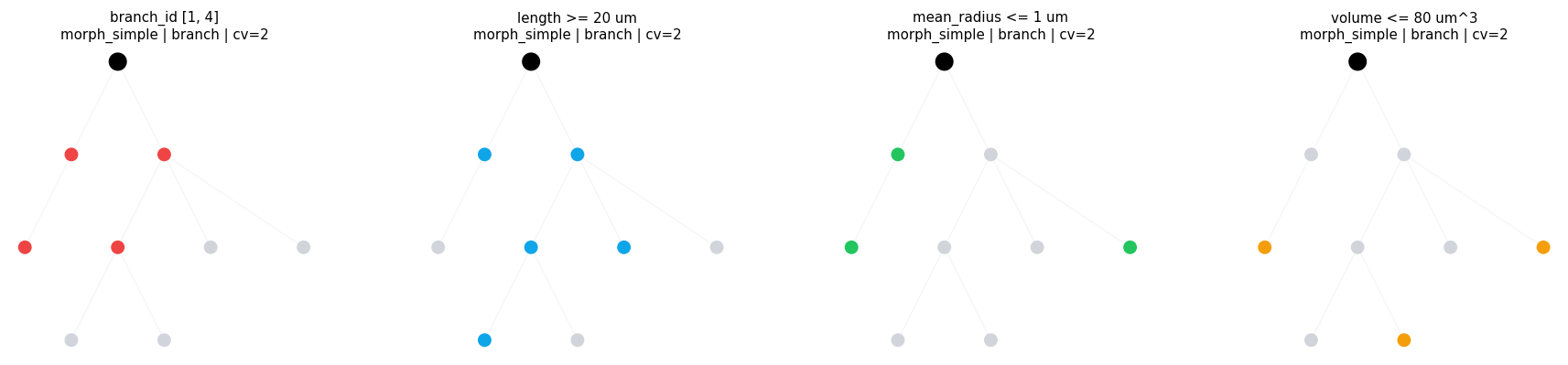
3.4 Region Set Algebra#
Region expressions compose with union, intersection, difference, and complement.
left = BranchSlice(branch_index=3, prox=0.00, dist=0.60)
right = BranchSlice(branch_index=3, prox=0.40, dist=1.00)
exclude = BranchSlice(branch_index=[1, 3], prox=[0.00, 0.20], dist=[1.00, 0.80])
compare_regions(
morph_pc,
[
('left | right', left | right),
('left & right', left & right),
('left - right', left - right),
('exclude.complement()', exclude.complement()),
],
level='cv',
layout='twopi',
cv_per_branch=2,
)
compare_regions defaults: morph=morph_pc level=cv layout=twopi cv_per_branch=2
left | right morph=morph_pc level=cv layout=twopi cv_per_branch=2 intervals= 1 sample=((3, 0.0, 1.0),)
left & right morph=morph_pc level=cv layout=twopi cv_per_branch=2 intervals= 1 sample=((3, 0.4, 0.6),)
left - right morph=morph_pc level=cv layout=twopi cv_per_branch=2 intervals= 1 sample=((3, 0.0, 0.4),)
exclude.complement() morph=morph_pc level=cv layout=twopi cv_per_branch=2 intervals= 461 sample=((0, 0.0, 1.0), (2, 0.0, 1.0), (3, 0.0, 0.2), (3, 0.8, 1.0))

3.5 Same Logic on morph_io#
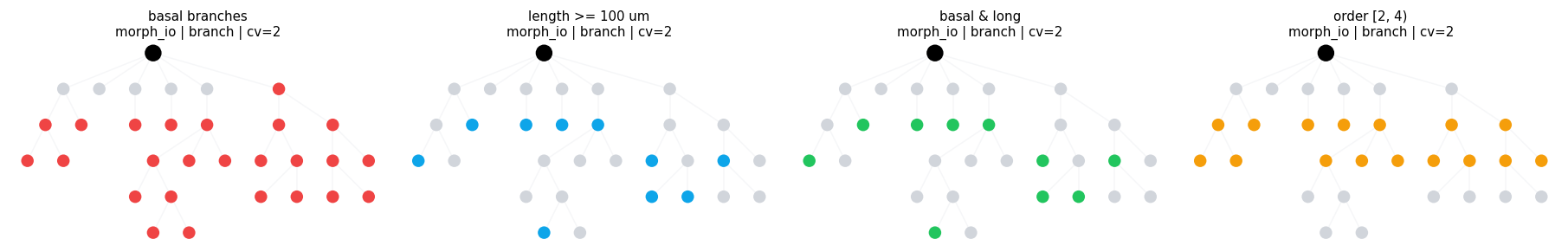
Once the filter expression is clear, the same calls work on a richer morphology.
basal = branch_in('type', 'basal_dendrite')
long = branch_range('length', (100.0 * u.um, None), closed='left')
mid_order = branch_range('branch_order', (2, 4), closed='left')
compare_regions(
morph_io,
[
('basal branches', basal),
('length >= 100 um', long),
('basal & long', basal & long),
('order [2, 4)', mid_order),
],
level='branch',
layout='dot',
)
compare_regions defaults: morph=morph_io level=branch layout=dot cv_per_branch=2
basal branches morph=morph_io level=branch layout=dot cv_per_branch=2 intervals= 25 sample=((6, 0.0, 1.0), (7, 0.0, 1.0), (8, 0.0, 1.0), (9, 0.0, 1.0))
length >= 100 um morph=morph_io level=branch layout=dot cv_per_branch=2 intervals= 10 sample=((8, 0.0, 1.0), (10, 0.0, 1.0), (11, 0.0, 1.0), (13, 0.0, 1.0))
basal & long morph=morph_io level=branch layout=dot cv_per_branch=2 intervals= 10 sample=((8, 0.0, 1.0), (10, 0.0, 1.0), (11, 0.0, 1.0), (13, 0.0, 1.0))
order [2, 4) morph=morph_io level=branch layout=dot cv_per_branch=2 intervals= 16 sample=((7, 0.0, 1.0), (8, 0.0, 1.0), (9, 0.0, 1.0), (12, 0.0, 1.0))
3.6 Same Logic on morph_pc#
On a larger morphology, use the same small vocabulary: metadata predicates, metric ranges, and set algebra.
deep = branch_range('branch_order', (6, 8), closed='left')
short = branch_range('length', (None, 5.0 * u.um), closed='right')
thin = branch_range('mean_radius', (None, 0.5 * u.um), closed='right')
compare_regions(
morph_pc,
[
('order [6, 8)', deep),
('length <= 5 um', short),
('mean_radius <= 0.5 um', thin),
('deep & short', deep & short),
],
level='branch',
layout='dot',
width=8.4,
height=10.0,
)
compare_regions defaults: morph=morph_pc level=branch layout=dot cv_per_branch=2
order [6, 8) morph=morph_pc level=branch layout=dot cv_per_branch=2 intervals= 52 sample=((6, 0.0, 1.0), (7, 0.0, 1.0), (8, 0.0, 1.0), (9, 0.0, 1.0))
length <= 5 um morph=morph_pc level=branch layout=dot cv_per_branch=2 intervals= 113 sample=((7, 0.0, 1.0), (8, 0.0, 1.0), (9, 0.0, 1.0), (10, 0.0, 1.0))
mean_radius <= 0.5 um morph=morph_pc level=branch layout=dot cv_per_branch=2 intervals= 328 sample=((5, 0.0, 1.0), (6, 0.0, 1.0), (7, 0.0, 1.0), (8, 0.0, 1.0))
deep & short morph=morph_pc level=branch layout=dot cv_per_branch=2 intervals= 18 sample=((7, 0.0, 1.0), (8, 0.0, 1.0), (9, 0.0, 1.0), (10, 0.0, 1.0))

4. Locset Filters#
A locset is a set of points: (branch_id, x).
Locsets are separate from regions because they mark point-process sites instead of continuous branch intervals.
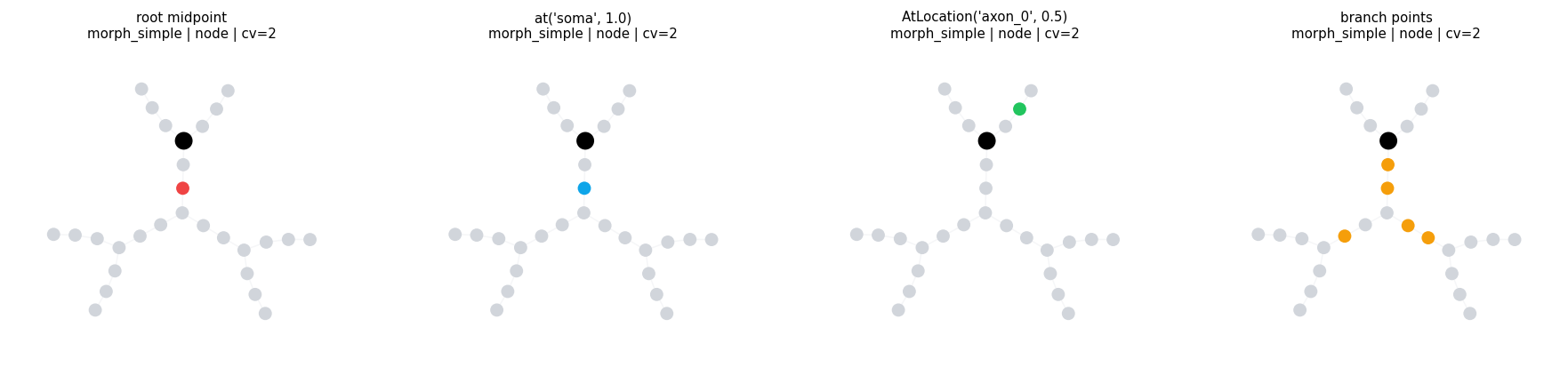
4.1 Direct Point Selectors#
Use direct locsets when you know the exact site or want built-in structural points.
compare_locsets(
morph_simple,
[
('root midpoint', RootLocation(x=0.5)),
("at('soma', 1.0)", at('soma', 1.0)),
("AtLocation('axon_0', 0.5)", AtLocation(branch='axon_0', x=0.5)),
('branch points', BranchPoints()),
],
level='node',
layout='kamada_kawai',
)
compare_locsets defaults: morph=morph_simple level=node layout=kamada_kawai cv_per_branch=2
root midpoint morph=morph_simple level=node layout=kamada_kawai cv_per_branch=2 points= 1 sample=('soma(0.5)',)
at('soma', 1.0) morph=morph_simple level=node layout=kamada_kawai cv_per_branch=2 points= 1 sample=('soma(1)',)
AtLocation('axon_0', 0.5) morph=morph_simple level=node layout=kamada_kawai cv_per_branch=2 points= 1 sample=('axon_0(0.5)',)
branch points morph=morph_simple level=node layout=kamada_kawai cv_per_branch=2 points= 5 sample=('soma(0)', 'soma(1)', 'basal_dendrite_0(0)', 'basal_dendrite_0(1)')
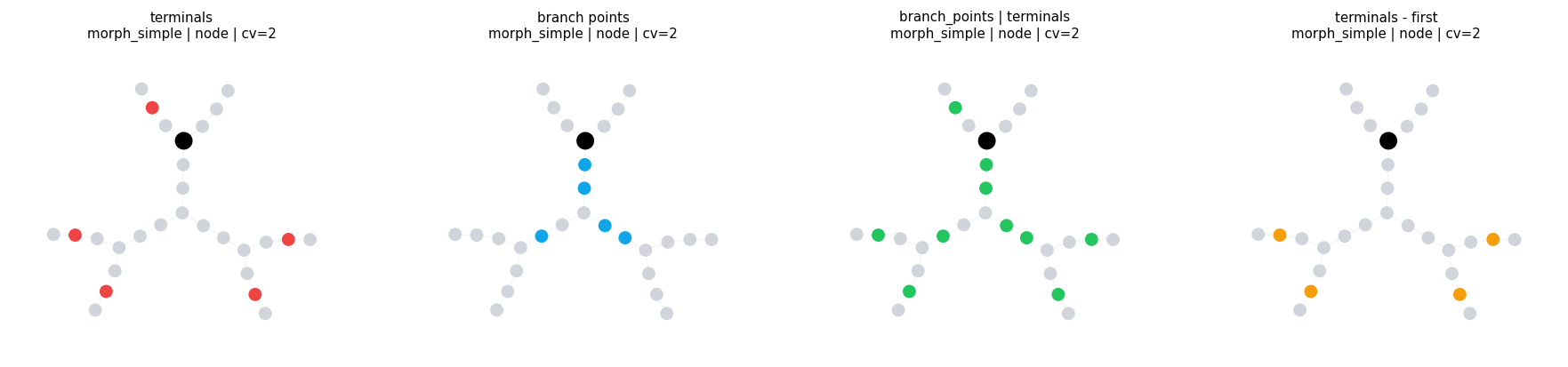
4.2 Structural Locsets and Set Algebra#
Locsets also compose with union, intersection, and difference.
branch_points = BranchPoints()
terminals = Terminals()
first_terminal = morph_simple.select(terminals).points[0]
compare_locsets(
morph_simple,
[
('terminals', terminals),
('branch points', branch_points),
('branch_points | terminals', branch_points | terminals),
('terminals - first', terminals - at(first_terminal[0], first_terminal[1])),
],
level='node',
layout='kamada_kawai',
)
compare_locsets defaults: morph=morph_simple level=node layout=kamada_kawai cv_per_branch=2
terminals morph=morph_simple level=node layout=kamada_kawai cv_per_branch=2 points= 5 sample=('axon_1(1)', 'basal_dendrite_2(1)', 'basal_dendrite_3(1)', 'basal_dendrite_4(1)')
branch points morph=morph_simple level=node layout=kamada_kawai cv_per_branch=2 points= 5 sample=('soma(0)', 'soma(1)', 'basal_dendrite_0(0)', 'basal_dendrite_0(1)')
branch_points | terminals morph=morph_simple level=node layout=kamada_kawai cv_per_branch=2 points= 10 sample=('soma(0)', 'soma(1)', 'axon_1(1)', 'basal_dendrite_0(0)')
terminals - first morph=morph_simple level=node layout=kamada_kawai cv_per_branch=2 points= 4 sample=('basal_dendrite_2(1)', 'basal_dendrite_3(1)', 'basal_dendrite_4(1)', 'basal_dendrite_5(1)')
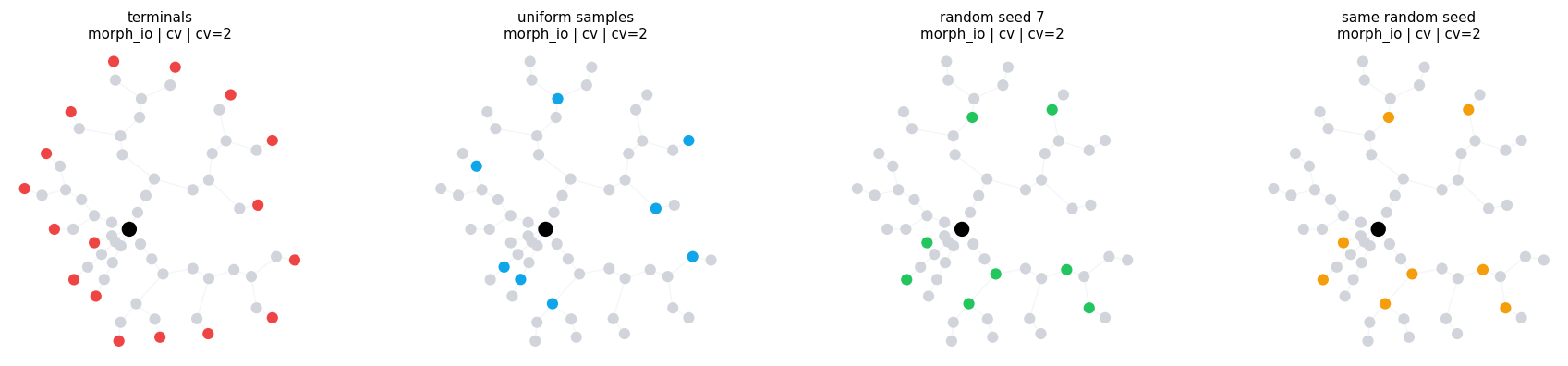
4.3 Sampling Points from a Region#
UniformSamples and RandomSamples take a region expression, then choose points inside it.
sampling_region = branch_range('branch_order', (1, None), closed='left')
uniform = UniformSamples(region=sampling_region, count=8)
random_a = RandomSamples(region=sampling_region, count=8, seed=7)
random_b = RandomSamples(region=sampling_region, count=8, seed=7)
print('same random seed gives same points:', morph_io.select(random_a).points == morph_io.select(random_b).points)
print()
compare_locsets(
morph_io,
[
('terminals', Terminals()),
('uniform samples', uniform),
('random seed 7', random_a),
('same random seed', random_b),
],
level='cv',
layout='twopi',
)
same random seed gives same points: True
compare_locsets defaults: morph=morph_io level=cv layout=twopi cv_per_branch=2
terminals morph=morph_io level=cv layout=twopi cv_per_branch=2 points= 17 sample=('soma_1(1)', 'basal_dendrite_2(1)', 'basal_dendrite_4(1)', 'basal_dendrite_5(1)')
uniform samples morph=morph_io level=cv layout=twopi cv_per_branch=2 points= 8 sample=('basal_dendrite_2(0.405)', 'basal_dendrite_4(0.537347)', 'basal_dendrite_7(0.548927)', 'basal_dendrite_12(0.435744)')
random seed 7 morph=morph_io level=cv layout=twopi cv_per_branch=2 points= 8 sample=('soma_1(0.504548)', 'basal_dendrite_5(0.278426)', 'basal_dendrite_7(0.25487)', 'basal_dendrite_15(0.797069)')
same random seed morph=morph_io level=cv layout=twopi cv_per_branch=2 points= 8 sample=('soma_1(0.504548)', 'basal_dendrite_5(0.278426)', 'basal_dendrite_7(0.25487)', 'basal_dendrite_15(0.797069)')
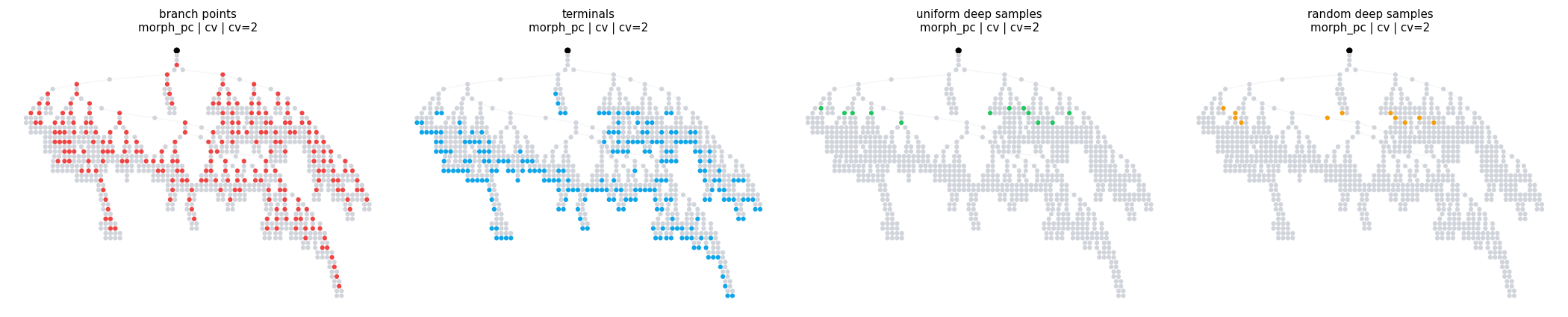
4.4 Locsets on morph_pc#
The same locset calls work on the larger morphology. The filter logic stays small; only the morphology changes.
pc_sampling_region = branch_range('branch_order', (6, 8), closed='left')
compare_locsets(
morph_pc,
[
('branch points', BranchPoints()),
('terminals', Terminals()),
('uniform deep samples', UniformSamples(region=pc_sampling_region, count=12)),
('random deep samples', RandomSamples(region=pc_sampling_region, count=12, seed=3)),
],
level='cv',
layout='dot',
width=4.4,
height=4.0,
)
compare_locsets defaults: morph=morph_pc level=cv layout=dot cv_per_branch=2
branch points morph=morph_pc level=cv layout=dot cv_per_branch=2 points= 228 sample=('dendrite_0(1)', 'dendrite_1(1)', 'dendrite_2(1)', 'dendrite_3(1)')
terminals morph=morph_pc level=cv layout=dot cv_per_branch=2 points= 229 sample=('dendrite_6(1)', 'dendrite_7(1)', 'dendrite_10(1)', 'dendrite_11(1)')
uniform deep samples morph=morph_pc level=cv layout=dot cv_per_branch=2 points= 12 sample=('dendrite_8(0.333287)', 'dendrite_20(0.845796)', 'dendrite_80(0.793563)', 'dendrite_88(0.921427)')
random deep samples morph=morph_pc level=cv layout=dot cv_per_branch=2 points= 12 sample=('dendrite_17(0.284201)', 'dendrite_17(0.430628)', 'dendrite_20(0.97346)', 'dendrite_26(0.292721)')
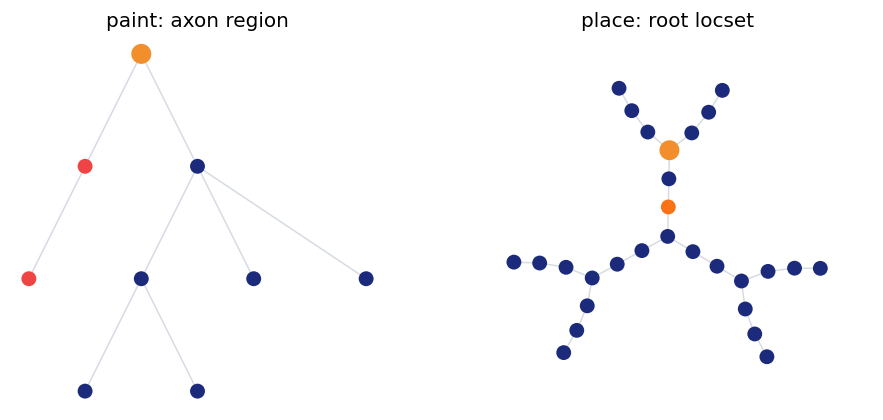
5. Filters as Cell Inputs#
The same expressions are the user-facing handles for mechanism placement.
cell = Cell(morph_simple, cv_policy=CVPerBranch(cv_per_branch=2))
axon_region = branch_in('type', 'axon')
probe_site = RootLocation(0.5)
cell.paint(
axon_region,
mech.Channel('IL', g_max=0.03 * (u.mS / u.cm ** 2), E=-65.0 * u.mV),
)
cell.place(probe_site, mech.StateProbe())
cell.init_state()
print(cell)
print('painted region intervals:', morph_simple.select(axon_region).intervals)
print('probe site points :', morph_simple.select(probe_site).display_names)
fig, axes = plt.subplots(1, 2, figsize=(8.0, 3.6))
cell.vis_topology(level='branch', layout='dot', region=axon_region, ax=axes[0], show=False)
axes[0].set_title('paint: axon region')
cell.vis_topology(level='node', layout='kamada_kawai', layout_scale=1.5, locset=probe_site, ax=axes[1], show=False, highlight_color='#f97316')
axes[1].set_title('place: root locset')
plt.tight_layout()
plt.show()
Cell(root='soma', n_cv=18, n_point=28, initialized=True)
painted region intervals: ((1, 0.0, 1.0), (2, 0.0, 1.0))
probe site points : ('soma(0.5)',)
6. Quick Reference#
Plot configuration lives in the global constants near the top of the notebook. You can also override per panel with dictionaries:
compare_regions(
morph_simple,
[
{'label': 'cv view', 'expr': expr, 'level': 'cv', 'layout': 'twopi', 'cv_per_branch': 2},
{'label': 'branch view', 'expr': expr, 'level': 'branch', 'layout': 'dot'},
],
)
Implemented region selectors:
API |
Use when |
|---|---|
|
select all / no branch intervals |
|
select explicit |
|
match branch metadata, topology, or scalar metric values |
|
filter numeric or |
`left |
right |
Supported scalar branch properties:
metadata / topology:
branch_id,name,type,parent_id,n_children,branch_order,n_tapersgeometry metrics:
length,mean_radius,area,volume
Implemented locset selectors:
API |
Use when |
|---|---|
|
choose a point on branch |
|
choose an explicit branch-local point |
|
choose branch-point sites on the parent side |
|
choose terminal branch distal ends |
|
evenly sample points from a region |
|
reproducibly sample random points from a region |
`left |
right |
Exported but not implemented yet: RadiusRangeRegion, TreeDistanceRegion, EuclideanDistanceRegion, SubtreeRegion, RegionAnchors, and StepSamples.